FDA telah meng-approved 20 obat pada tahun 2010. Obat-obat tersebut
adalah hampir sama efek farmakologisnya dengan obat yang sudah
dipasarkan (Tabel 1). Ada 2 orphan drug (obat untuk kasus langka) yaitu
untuk penyakit Dupuytren dan defisiensi n-acetylglutamate synthase
(Tabel 2). Ada obat “first in class” yaitu untuk kontrasepsi emergensi, lipodystrophy,
multiple sclerosis, and rheumatoid arthritis (Tabel 3). Satu obat baru
yaitu eribulin, suatu antimitotik yang diindikasikan untuk kanker
payudara, suatu obat yang
diperoleh dari sponge laut, Halichondria okadai, obat yang dipasarkan ketiga yang berasal dari marine invertebrates (buka referensi ini untuk mendiskusikan tantangan memanen obat dari laut).
Obsolete Indications:
Market Withdrawals/Suspensions:
Update on the Clinical Impact of USP Heparin Potency Reduction: FDA has clarified that the 10% decrease in anticoagulation activity of heparin products manufactured under the new United States Pharmacopeia (USP) standard adopted in 2009, and originally presumed to be clinically insignificant, may warrant adjustments in heparin dosage and/or more intensive monitoring, particularly in the following situations: (1) extracorporeal membrane oxygenation in pediatric patients, (2) cardiopulmonary bypass, and (3) treatment or prevention of life-threatening thromboses.
“Repurposed Drugs”: Four established drugs were approved for new purposes or as novel formulations for established purposes (see Table 4 ).
In addition, FDA granted the following significant vaccine and new biologic licenses in 2010:
Collagenase clostridium histolyticum (XIAFLEX) consists of a 1:1 ratio of two microbial collagenases isolated and from the fermentation of Clostridium histolyticum bacteria.
Collagenase AUX-I is a class I collagenase that preferentially cleaves
the ends of collagen molecules and collagenase AUX-II is a class II
collagenase that preferentially cleaves the center portions of collagen
molecules.[3] The drug was granted orphan drug status
in the United States for Dupuytren disease in 1996. FDA approval in 2010
was based on the efficacy demonstrated during two randomized,
placebo-controlled, multicenter trials involving 374 patients. Dupuytren
disease is a condition in which the tendons of the hand that move the
fingers become thickened and scarred by collagen deposits causing a
fixed flexion deformity.[4] When injected into a
palpable Dupuytren disease cord, collagenase clostridium histolyticum
hydrolyzes the collagen molecules in the cord resulting in enzymatic
rupture. A follow-on office visit for a finger extension manipulation is
usually necessary to reduce the contracture and daily home exercise is
necessary to regain and retain range of motion following treatment. Up
to three repeat injections per cord may be administered if needed. The
enzymatic alternative appears to compare favorably to surgical
fasciectomy/fasciotomy. Collagenase clostridium histolyticum must be
administered with caution by a healthcare provider experienced with the
injection procedure because enzymatic exposure external to the cord may
result in permanent injury of non-targeted tissues (e.g., tendons,
nerves, blood vessels). During pre-market clinical trials, at least 3
tendon ruptures were reported. Other common side effects of collagenase
clostridium histolyticum are pain, swelling, bruising, and bleeding at
the injection site. Foreign-protein hypersensitivity reactions are to be
expected following repeat injections. In addition, patients are at risk
of fainting during the post-injection finger extension procedure.
Fingolimod (Figure 2)(GILENYA) is the first oral medication for multiple sclerosis to be marketed in the United States. Fingolimod is indicated to reduce the frequency of clinical exacerbations and to delay physical disability in patients with multiple sclerosis. The drug demonstrated superiority over interferon beta-1a (AVONEX, REBIF) in both outcomes over the course of 12 months of study.[7] Following oral administration, fingolimod is metabolized by sphingosine kinase to the active metabolite, fingolimod-phosphate. Fingolimod-phosphate is a non-specific sphingosine 1-phosphate (S1P) receptor modulator. With five S1P receptors characterized, the pharmacological actions of fingolimod-phosphate are predictably complex. For patients with multiple sclerosis, the main therapeutic benefits are thought to involve reducing the migration of lymphocytes into the central nervous system (CNS) by blocking their egress from lymph nodes. Besides this regulation of lymphocyte “trafficking,” S1P receptors also are thought to play a role in the regulation of angiogenesis, neurogenesis, smooth-muscle contraction, heart rate, endothelial barrier integrity, and vascular tone (S1P-1 receptor); auditory and vestibular functioning and neuronal excitability (S1P-2 receptor); heart rate (S1P-3 receptor); smooth muscle cells lining the airways (S1P-4 receptor); and oligodendrocyte functioning in the white matter of the CNS (S1P-5 receptor).[8,9]
Consistent with the broad range of actions attributed to S1P
receptors, fingolimod is associated a number of undesired effects which
include: bradycardia (mean 13 beat/minute decrease in heart rate
following the first dose), heart block, immunosuppression, lymphoma,
serious infection, macular edema, change in visual acuity, reduction in
pulmonary function, hepatic dysfunction, and hypertension. In addition
to these risks, patients receiving fingolimod must be monitored for
hepatotoxicity and women of childbearing potential should use effective
contraception to avoid pregnancy during, and extending for 2 months
after stopping, therapy with fingolimod. Untoward reactions may also
occur in patients receiving antiarrhythmic drugs, beta-blockers, calcium
channel blockers, antihypertensive drugs, drugs for heart failure,
ketoconazole, live attenuated vaccines, antineoplastics,
immunosuppressives, immune modulators, and antigenic testing
concurrently with fingolimod and for up to 2 months after its
discontinuation.
The recommended dose of fingolimod is 0.5 mg orally once daily. Patients should have baseline laboratories drawn before starting therapy and should be monitored for bradycardia for 6 hours after the first dose. Fingolimod is well absorbed orally with the Tmax delayed until 12-16 hours after a dose. Fingolimod is extensively distributed to body tissues with a volume of distribution of approximately 1200 ± 260 liters. Fingolimod and fingolimod-phosphate are >99% protein bound. Steady-state concentrations (approximately 10-fold greater than after the initial dose) are reached within 1 to 2 months. The half-life for elimination of fingolimod averages 6-9 days; however, the pharmacodynamic effects (including decreased lymphocyte counts) persist for up to 2 months. Fingolimod is mainly excreted in the urine as inactive metabolites. There are three pathways involved in the metabolism of fingolimod: (1) phosphorylation to the pharmacologically active fingolimod-phosphate; (2) biotransformation mainly by cytochrome P450 isozyme 4F2 (but also 2D6, 2E1, 3A4, and 4F12) with subsequent degradation to inactive metabolites; and (3) formation of inactive ceramide analogs.
Tesamorelin (Figure 3) (EGRIFTA) (PubChem # 44201342) is an analog of growth hormone-releasing hormone (GHRH) approved for reducing visceral adipose tissue in patients with HIV-associated lipohypertrophy. Tesamorelin is administered subcutaneously once daily. The drug is contraindicated in patients with disruption of the hypothalamic-pituitary axis, active malignancy, and during pregnancy (FDA pregnancy category X). Adverse effects include fluid retention, hemoglobin A1c elevation, glucose intolerance, development of diabetes, injection site reactions, and hypersensitivity reactions. Approximately 50% of patients develop anti-tesamorelin IgG antibodies and 5-10% of patients develop tesamorelin-neutralizing antibodies. Approximately 60% of anti-tesamorelin IgG antibodies are cross-reactive with endogenous growth hormone-releasing hormone. The pharmacologic effect of tesamorelin is to induce growth hormone (GH) secretion from the pituitary and secondarily to result in insulin-like growth hormone 1 (IGF-1) secretion from the liver and peripheral tissues. All of the clinical considerations for the administration of exogenous growth hormone apply. By extension, patients who are critically ill at are risk for increased mortality due to the pharmacologic effects of growth hormone, and may experience altered clearance of compounds metabolized by CYP450 liver isozymes. Careful monitoring of patients receiving concomitant therapy with CYP450 substrates is warranted, particularly patients receiving chronic corticosteroids and patients with hypoadrenalism receiving glucocorticoid replacement therapy. FDA approval of tesamorelin was on the basis of the modest (14-18%) reduction in visceral adipose tissue by week 26 of treatment. On average the reduction was demonstrated to be sustainable for up to a year of treatment with regain of the fat lost upon discontinuation of therapy. Pre-market studies were not robust enough to determine the long-term implications for cardiovascular risk in the population studied. The magnitude of fat loss with tesamorelin is similar to the results attainable through diet and exercise.[10] Given the potential for adverse effects, patients receiving tesamorelin require careful monitoring, particularly for altered glucose metabolism.
Tocilizumab (ACTEMRA) is the 9th immunomodulator to be FDA approved for rheumatoid arthritis.[11]
Tocilizumab is the first-in-class anti-interleukin 6 (IL-6) receptor
monoclonal antibody. It has been available for use in Japan since 2005.[11]
IL-6 is a pleiotropic pro-inflammatory cytokine produced by T- and
B-cells, lymphocytes, monocytes, fibroblasts, and synovial and
endothelial cells of joints affected by rheumatoid arthritis.[11]
Of importance, IL-6 induces osteoclast differentiation and can be
responsible for joint destruction in patients suffering from rheumatoid
arthritis.[11] Tocilizumab inhibits signaling through both forms of the IL-6 receptor, sIL-6R and mIL-6R.[12]
Tocilizumab is specifically indicated for once-a-month intravenous
infusion, with or without methotrexate, for patients who have failed
therapy with one or more TNF antagonists. Tocilizumab shares the adverse
effect profile of other monoclonal immunosuppressors, including
infusion reactions, development of neutralizing antibodies,
hypersensitivity reactions, elevation of the risk for serious infections
and reactivation of latent infections, and the potential for
development of malignancy. Laboratory monitoring of neutrophils,
platelets, lipids, and liver function is necessary and the dose of
tocilizumab must be adjusted on the basis of abnormal findings.
Hyperlipidemia, gastrointestinal perforation, and symptoms of
demyelinating disorders have been associated with tocilizumab therapy.
Tocilizumab may result in a shift in the expression of hepatic CYP450
enzymes and may lead to clinically significant drug interactions with
narrow therapeutic index substrates of these isozymes. The onset of
action of tocilizumab is heralded by a decrease in levels of C-reactive
protein within 2 weeks of the first dose. Changes in other
pharmacodynamic parameters include decreases in rheumatoid factor,
erythrocyte sedimentation rate, and serum amyloid A as well as increases
in hemoglobin. The half-life for tocilizumab elimination is 11-12 days.
The efficacy of tocilizumab was evaluated in five controlled studies of
patients who had an inadequate clinical response to methotrexate, other
disease-modifying antirheumatic drugs, or TNF antagonist therapy.
Efficacy was measured as the percentage of patients achieving a 20%
improvement in disease activity according to American College of
Rheumatology criteria.[12] While the overall response
rate to tocilizumab appears to be similar to other biologics,
tocilizumab is the first biologic for rheumatoid arthritis to
demonstrate head-to-head superiority over methotrexate.[12]
Also similar to trials of other biologics, the response to tocilizumab
trended lower with prior disease duration and prior therapeutic
experience.[12]
Ulipristal (Figure 4) (ELLA) is the first progesterone receptor modulator with antagonist and partial agonist effects. Ulipristal is approved for use as a 30 mg single-dose emergency contraceptive. Unlike levonorgestrel-based emergency contraceptives (PLAN B, PLAN B ONE STEP, NEXT CHOICE) that are available over-the-counter and must be taken within 3 days of unprotected intercourse, ulipristal is only available by prescription, but can be taken up to 5 days after unprotected intercourse. The most common adverse reactions following ulipristal are headache (18%), abdominal pain (12%), nausea (12%), dysmenorrhea (9%), fatigue (6%), dizziness (5%), and a one-time disruption to the menstrual cycle length. Due to its high affinity for the progesterone receptor, ulipristal may reduce the efficacy of regular hormonal contraceptives necessitating supplemental use of a reliable barrier contraceptive method until the onset of menses.[13] Predominantly metabolized by CYP3A4 to mono- and di-demethylated metabolites, ulipristal also has the potential for drug-drug interactions with compounds that induce or inhibit CYP3A4. Like other progesterone agonists, the pharmacological actions of ulipristal are specific to the phase of the menstrual cycle.[13] When taken immediately before ovulation, ulipristal postpones follicular rupture. Therefore, the most likely mechanism of action of ulipristal for emergency contraception is ovulation delay or inhibition.
diperoleh dari sponge laut, Halichondria okadai, obat yang dipasarkan ketiga yang berasal dari marine invertebrates (buka referensi ini untuk mendiskusikan tantangan memanen obat dari laut).
Table 1. New Drugs Licensed in 2010 with Pharmacological Mechanisms Similar to Previously Approved Drugs
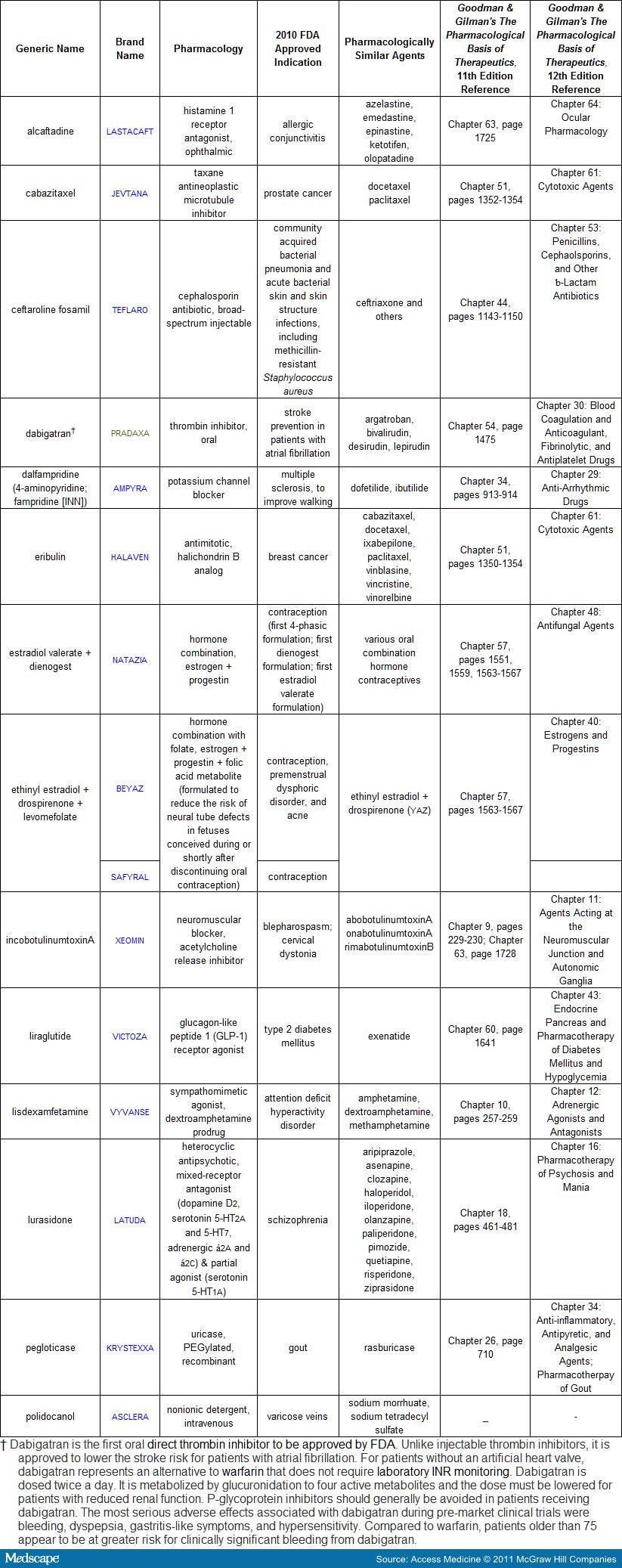
Table 2. Orphan Drugs Granted FDA Approval in 2010 (also see monographs below)

Table 3. New Pharmacological Drug Classes Introduced in 2010 (also see monographs below)
Perubahan Brand Name: Brand name untuk dexlansoprazole telah diubah dari KAPIDEX menjadi DEXILANT. Hal ini dilakukan untuk menghindari kerancuan nama dengan CASODEX (bicalutamide).

Obsolete Indications:
- Bevacizumab (AVASTIN) does not appear to prolong survival, nor provide sufficient benefit in slowing disease progression, to outweigh the risk to patients with metastatic breast cancer and FDA has proposed withdrawing this (one) indication for the drug. A public hearing to provide FDA with additional evidence to confirm that the drug is safe and effective for this indication has been requested.
- Dolasetron (ANZEMET) dapat meningkatkan torsades de pointes dan hal ini tidak direkomendasikan untuk pengobatan mual dan muntah yang terinduksi kemoterapi.
- Quinine (QUALAQUIN) can cause life-threatening thrombocytopenia, hemolytic-uremic syndrome, and thrombotic thrombocytopenic purpura. Quinine is no longer recommended for the off-label use for leg cramps
Market Withdrawals/Suspensions:
- Gemtuzumab (MYLOTARG), a monoclonal antibody (specific for the CD33 antigen) and cytotoxic conjugate, was voluntarily withdrawn from the United States market due to a lack of post-marketing confirmation of a survival benefit for patients with acute myeloid leukemia . Generic manufacturers have been informed of FDA’s recommendation for healthcare professionals to stop prescribing the drug, but it may be some time before the drug is completely purged from the United States market.
- Propoxyphene (DARVON, DARVOCET), FDA approved since 1957, was withdrawn from the United States market due to evidence of risk for serious heart rhythm abnormalities (see Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edition Chapter 21, pages 573-574; 12th edition, Chapter 18).
- Rosiglitazone (AVANDIA, AVANDAMET, AVAGLIM) has been suspended from marketing in Europe and placed on a restricted distribution status in the United States[2] due to post-marketing detection of signals of increased cardiovascular risk
- Sibutramine (MERIDIA) secara sukarela ditarik dari Amerika Serikat karena pada post-marketing terdeteksi meningkatkan risiko serangan jantung dan stroke yang dihubungkan dengan penggunaan obat ini untuk menurunkan berat badan.
Update on the Clinical Impact of USP Heparin Potency Reduction: FDA has clarified that the 10% decrease in anticoagulation activity of heparin products manufactured under the new United States Pharmacopeia (USP) standard adopted in 2009, and originally presumed to be clinically insignificant, may warrant adjustments in heparin dosage and/or more intensive monitoring, particularly in the following situations: (1) extracorporeal membrane oxygenation in pediatric patients, (2) cardiopulmonary bypass, and (3) treatment or prevention of life-threatening thromboses.
“Repurposed Drugs”: Four established drugs were approved for new purposes or as novel formulations for established purposes (see Table 4 ).
In addition, FDA granted the following significant vaccine and new biologic licenses in 2010:
- A photodynamic imaging agent, hexaminolevulinate hydrochloride (CYSVIEW), was licensed for the detection of superficial bladder cancer during cystoscopy.
- Quadrivalent human papillomavirus (HPV) vaccine (Gardasil) licensure was extended for children and young adults aged 9 to 26 years to include the prevention of anal cancer and associated precancerous lesions due to HPV types 6, 11, 16, and 18.
- A fourth alpha1-proteinase inhibitor (Glassia) was licensed for therapy in patients with emphysema caused by a congenital deficiency of alpha1-antitrypsin.
- A new 13-valent pneumococcal conjugate vaccine (Prevnar 13) was licensed as the successor to the 7-valent vaccine, Prevnar (licensed in 2000), to prevent invasive pneumococcal disease and otitis media in children aged 6-weeks to 5-years old.
- The first RANK ligand inhibitor, denosumab (PROLIA, XGEVA), was licensed for prevention of skeletal fractures and pain in patients with bone metastases from solid tumors (XGEVA) and for the treatment of postmenopausal osteoporosis (PROLIA) (see Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edition Chapter 61, page 1658 and figure 16-9, page 1659; 12th edition, Chapter 44).
- An autologous cellular immunotherapy, sipuleucel-T (PROVENGE) was licensed for the treatment of metastatic hormone-refractory prostate cancer. The therapy consists of administering autologous prostatic acid phosphatase-presenting cells which have been activated in vitro by granulocyte-macrophage colony-stimulating factor (see Goodman & Gilman’s The Pharmacological Basis of Therapeutics, 11th edition Chapter 53, pages 1439-1440; 12th edition, Chapter 37).
- An absorbable fibrin sealant patch (TACHOSIL) was licensed for use as an adjunct to hemostasis in cardiovascular surgery when control of bleeding by suture, ligature or cautery, is ineffective or impractical.
- Velaglucerase alfa (VPRIV), a glucocerebroside-specific enzyme, was licensed for long-term replacement therapy for patients with type 1 Gaucher disease.
(Enlarge Image)
|
Figure 1. Chemical structure of carglumic acid (PubChem CID: 121396) compared to n-acetylglutamate (PubChem CID: 70914) |
Fingolimod (Figure 2)(GILENYA) is the first oral medication for multiple sclerosis to be marketed in the United States. Fingolimod is indicated to reduce the frequency of clinical exacerbations and to delay physical disability in patients with multiple sclerosis. The drug demonstrated superiority over interferon beta-1a (AVONEX, REBIF) in both outcomes over the course of 12 months of study.[7] Following oral administration, fingolimod is metabolized by sphingosine kinase to the active metabolite, fingolimod-phosphate. Fingolimod-phosphate is a non-specific sphingosine 1-phosphate (S1P) receptor modulator. With five S1P receptors characterized, the pharmacological actions of fingolimod-phosphate are predictably complex. For patients with multiple sclerosis, the main therapeutic benefits are thought to involve reducing the migration of lymphocytes into the central nervous system (CNS) by blocking their egress from lymph nodes. Besides this regulation of lymphocyte “trafficking,” S1P receptors also are thought to play a role in the regulation of angiogenesis, neurogenesis, smooth-muscle contraction, heart rate, endothelial barrier integrity, and vascular tone (S1P-1 receptor); auditory and vestibular functioning and neuronal excitability (S1P-2 receptor); heart rate (S1P-3 receptor); smooth muscle cells lining the airways (S1P-4 receptor); and oligodendrocyte functioning in the white matter of the CNS (S1P-5 receptor).[8,9]
(Enlarge Image)
|
Figure 2. Chemical structure of fingolimod (PubChem CID: 107969) |
The recommended dose of fingolimod is 0.5 mg orally once daily. Patients should have baseline laboratories drawn before starting therapy and should be monitored for bradycardia for 6 hours after the first dose. Fingolimod is well absorbed orally with the Tmax delayed until 12-16 hours after a dose. Fingolimod is extensively distributed to body tissues with a volume of distribution of approximately 1200 ± 260 liters. Fingolimod and fingolimod-phosphate are >99% protein bound. Steady-state concentrations (approximately 10-fold greater than after the initial dose) are reached within 1 to 2 months. The half-life for elimination of fingolimod averages 6-9 days; however, the pharmacodynamic effects (including decreased lymphocyte counts) persist for up to 2 months. Fingolimod is mainly excreted in the urine as inactive metabolites. There are three pathways involved in the metabolism of fingolimod: (1) phosphorylation to the pharmacologically active fingolimod-phosphate; (2) biotransformation mainly by cytochrome P450 isozyme 4F2 (but also 2D6, 2E1, 3A4, and 4F12) with subsequent degradation to inactive metabolites; and (3) formation of inactive ceramide analogs.
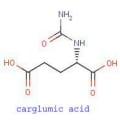
Tesamorelin (Figure 3) (EGRIFTA) (PubChem # 44201342) is an analog of growth hormone-releasing hormone (GHRH) approved for reducing visceral adipose tissue in patients with HIV-associated lipohypertrophy. Tesamorelin is administered subcutaneously once daily. The drug is contraindicated in patients with disruption of the hypothalamic-pituitary axis, active malignancy, and during pregnancy (FDA pregnancy category X). Adverse effects include fluid retention, hemoglobin A1c elevation, glucose intolerance, development of diabetes, injection site reactions, and hypersensitivity reactions. Approximately 50% of patients develop anti-tesamorelin IgG antibodies and 5-10% of patients develop tesamorelin-neutralizing antibodies. Approximately 60% of anti-tesamorelin IgG antibodies are cross-reactive with endogenous growth hormone-releasing hormone. The pharmacologic effect of tesamorelin is to induce growth hormone (GH) secretion from the pituitary and secondarily to result in insulin-like growth hormone 1 (IGF-1) secretion from the liver and peripheral tissues. All of the clinical considerations for the administration of exogenous growth hormone apply. By extension, patients who are critically ill at are risk for increased mortality due to the pharmacologic effects of growth hormone, and may experience altered clearance of compounds metabolized by CYP450 liver isozymes. Careful monitoring of patients receiving concomitant therapy with CYP450 substrates is warranted, particularly patients receiving chronic corticosteroids and patients with hypoadrenalism receiving glucocorticoid replacement therapy. FDA approval of tesamorelin was on the basis of the modest (14-18%) reduction in visceral adipose tissue by week 26 of treatment. On average the reduction was demonstrated to be sustainable for up to a year of treatment with regain of the fat lost upon discontinuation of therapy. Pre-market studies were not robust enough to determine the long-term implications for cardiovascular risk in the population studied. The magnitude of fat loss with tesamorelin is similar to the results attainable through diet and exercise.[10] Given the potential for adverse effects, patients receiving tesamorelin require careful monitoring, particularly for altered glucose metabolism.
(Enlarge Image)
|
Figure 3. |
Ulipristal (Figure 4) (ELLA) is the first progesterone receptor modulator with antagonist and partial agonist effects. Ulipristal is approved for use as a 30 mg single-dose emergency contraceptive. Unlike levonorgestrel-based emergency contraceptives (PLAN B, PLAN B ONE STEP, NEXT CHOICE) that are available over-the-counter and must be taken within 3 days of unprotected intercourse, ulipristal is only available by prescription, but can be taken up to 5 days after unprotected intercourse. The most common adverse reactions following ulipristal are headache (18%), abdominal pain (12%), nausea (12%), dysmenorrhea (9%), fatigue (6%), dizziness (5%), and a one-time disruption to the menstrual cycle length. Due to its high affinity for the progesterone receptor, ulipristal may reduce the efficacy of regular hormonal contraceptives necessitating supplemental use of a reliable barrier contraceptive method until the onset of menses.[13] Predominantly metabolized by CYP3A4 to mono- and di-demethylated metabolites, ulipristal also has the potential for drug-drug interactions with compounds that induce or inhibit CYP3A4. Like other progesterone agonists, the pharmacological actions of ulipristal are specific to the phase of the menstrual cycle.[13] When taken immediately before ovulation, ulipristal postpones follicular rupture. Therefore, the most likely mechanism of action of ulipristal for emergency contraception is ovulation delay or inhibition.


0 komentar:
Post a Comment